
SAE0022
Phosphoglucomutase 1 human
lyophilized powder, recombinant, expressed in E. coli
Sign Into View Organizational & Contract Pricing
All Photos(1)
About This Item
Recommended Products





recombinant
expressed in E. coli
Quality Level
description
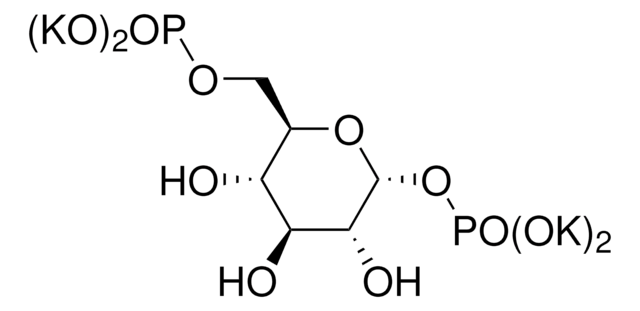
PGM1 isoform sequence with a predicted molecular mass of 61.3kDa.
Assay
≥95% (PAGE)
form
lyophilized powder
specific activity
≥400 units/mg protein
shipped in
dry ice
storage temp.
−20°C
General description
Phosphoglucomutase-1 (PGM1) is an evolutionarily conserved enzyme that belongs to the phosphohexose mutase family. It is the major isoform of PGM in skeletal muscle and most other tissues. The PGM1 gene is mapped to human chromosome 1p31.3.
Biochem/physiol Actions
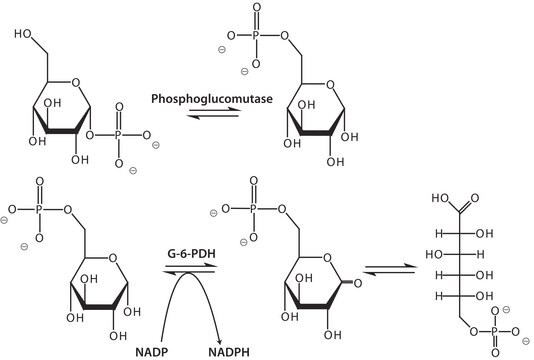
Phosphoglucomutase-1 (PGM1) catalyzes the bidirectional interconversion of glucose-1-phosphate (G-1-P) and glucose-6-phosphate (G-6-P). It regulates carbohydrate metabolism, energy production, and protein N-glycosylation. PGM1 participates in the biosynthesis of nucleotide sugars required for glycan biosynthesis. Variation in the PGM1 gene leads to PGM1 deficiency, which is considered an inherited metabolic disorder in humans. PGM1 deficiency causes autosomal recessive diseases such as glycogen storage disease type XIV and congenital disorder of protein N-glycosylation. Affected patients show multiple disease phenotypes, reflecting the central role of the enzyme in glucose homeostasis. The influence of PGM1 deficiency on protein glycosylation patterns is also widespread. PGM1 acts as a metabolic tumor suppressor.
Unit Definition
One unit will convert 1.0 μmole of α-D-Glucose-1-Phosphate to α-D-Glucose-6-phosphate per minute at pH 7.4 at 30 °C.
Physical form
Supplied as a lyophilized powder containing tris-buffered saline, EDTA, DTT and stabilizer.
Storage Class Code
11 - Combustible Solids
WGK
WGK 2
Flash Point(F)
Not applicable
Flash Point(C)
Not applicable
Certificates of Analysis (COA)
Search for Certificates of Analysis (COA) by entering the products Lot/Batch Number. Lot and Batch Numbers can be found on a product’s label following the words ‘Lot’ or ‘Batch’.
Already Own This Product?
Find documentation for the products that you have recently purchased in the Document Library.
Paul Hoff Backe et al.
Scientific reports, 10(1), 5656-5656 (2020-03-30)
Human phosphoglucomutase 1 (PGM1) is an evolutionary conserved enzyme that belongs to the ubiquitous and ancient α-D-phosphohexomutases, a large enzyme superfamily with members in all three domains of life. PGM1 catalyzes the bi-directional interconversion between α-D-glucose 1-phosphate (G1P) and α-D-glucose
Federica Conte et al.
Molecular genetics and metabolism, 131(1-2), 135-146 (2020-12-22)
Phosphoglucomutase 1 deficiency is a congenital disorder of glycosylation (CDG) with multiorgan involvement affecting carbohydrate metabolism, N-glycosylation and energy production. The metabolic management consists of dietary D-galactose supplementation that ameliorates hypoglycemia, hepatic dysfunction, endocrine anomalies and growth delay. Previous studies
Guang-Zhi Jin et al.
PLoS biology, 16(10), e2006483-e2006483 (2018-10-20)
Glycogen metabolism commonly altered in cancer is just beginning to be understood. Phosphoglucomutase 1 (PGM1), the first enzyme in glycogenesis that catalyzes the reversible conversion between glucose 1-phosphate (G-1-P) and glucose 6-phosphate (G-6-P), participates in both the breakdown and synthesis
Eva Morava
Molecular genetics and metabolism, 112(4), 275-279 (2014-07-07)
We recently redefined phosphoglucomutase-1 deficiency not only as an enzyme defect, involved in normal glycogen metabolism, but also an inborn error of protein glycosylation. Phosphoglucomutase-1 is a key enzyme in glycolysis and glycogenesis by catalyzing in the bidirectional transfer of
Wo-Tu Tian et al.
Neuromuscular disorders : NMD, 29(4), 282-289 (2019-02-10)
The congenital disorders of glycosylation are a group of clinically and biochemically heterogeneous diseases characterized by multisystem involvement due to glycosylation defect of protein and lipid. Here we report a 49-year-old man with exercise-induced fatigue and pain of muscle, tachypnea
Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.
Contact Technical Service