
43659
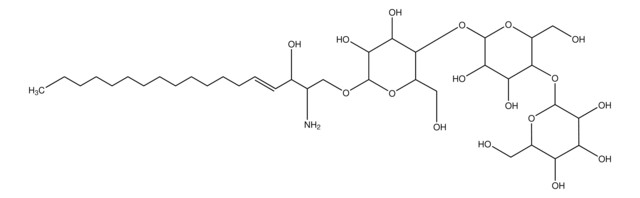
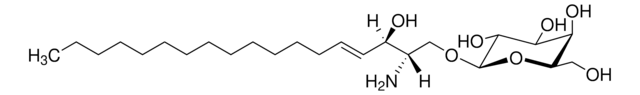
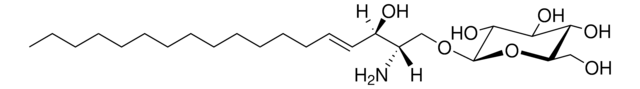
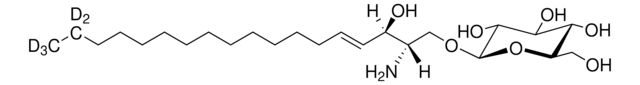
Glucosylsphingosine
≥98.0% (TLC)
동의어(들):
(2S,3R,4E)-2-Amino-3-hydroxy-4-octadecen-1-yl β-D-glucopyranoside, 1-β-D-Glucosylsphingosine, Glucosyl-C18-sphingosine
로그인조직 및 계약 가격 보기
모든 사진(1)
About This Item
실험식(Hill 표기법):
C24H47NO7
CAS Number:
Molecular Weight:
461.63
Beilstein:
4333674
MDL number:
UNSPSC 코드:
12352211
PubChem Substance ID:
NACRES:
NA.85
추천 제품

Quality Level
분석
≥98.0% (TLC)
양식
powder
지질 유형
sphingolipids
저장 온도
−20°C
SMILES string
O[C@@H]1[C@@H](O)[C@H](OC[C@H](N)[C@H](O)/C=C/CCCCCCCCCCCCC)O[C@H](CO)[C@H]1O
InChI
1S/C24H47NO7/c1-2-3-4-5-6-7-8-9-10-11-12-13-14-15-19(27)18(25)17-31-24-23(30)22(29)21(28)20(16-26)32-24/h14-15,18-24,26-30H,2-13,16-17,25H2,1H3/b15-14+/t18-,19+,20?,21+,22+,23?,24+/m0/s1
InChI key
HHJTWTPUPVQKNA-JLRUQHRASA-N
생화학적/생리학적 작용
Glucosylsphingosine is a cytotoxic compound. Accumulation of glucosylsphingosine in brain and other tissues occurs in patients with Gaucher disease, which is an inherited deficiency of lysosomal glucocerebrosidase, which converts glucosylsphingosine to glucose and sphingosine.
Storage Class Code
11 - Combustible Solids
WGK
WGK 3
Flash Point (°F)
Not applicable
Flash Point (°C)
Not applicable
이미 열람한 고객

N G Conradi et al.
Acta neuropathologica, 75(4), 385-390 (1988-01-01)
Splenectomy in children with the Norrbottnian type of Gaucher disease is followed by increased blood levels of glucosylceramide and impaired neurological and mental status. High blood levels are associated with an increased accumulation of glucosylceramide in perivascular Gaucher cells in
A Kaloterakis et al.
Journal of internal medicine, 246(6), 587-590 (2000-01-05)
Chronic Gaucher disease [GD] in association with systemic AL amyloidosis is extremely rare. We describe a 46-year-old Greek male with chronic GD confirmed by low glucocerebroside activity in fibroblasts and N370S/L444P mutations at the cerebrosidase gene, who also had systemic
Lulu Kang et al.
Journal of human genetics, 62(8), 763-768 (2017-03-31)
Gaucher disease (GD) is an inherited metabolic disorder that involves accumulation of glycolipid glucocerebroside in monocyte-macrophage cells, which can result in multiple organ damage. Enzyme replacement and substrate reduction therapies have improved the potential for early diagnosis and treatment. Determining
E Beutler
Blood reviews, 2(1), 59-70 (1988-03-01)
Gaucher disease is a glycolipid storage disorder characterized by accumulation of glucocerebroside in the liver, spleen, and bones, and caused by a deficiency of glucocerebrosidase. Glucocerebrosidase cDNA has been cloned and sequenced, and much has been learned about the synthesis
E M Kaye et al.
Annals of neurology, 20(2), 223-230 (1986-08-01)
Glucocerebroside levels were measured in the brains of patients with neuronopathic forms (types 2 and 3) of Gaucher disease and compared to those obtained from control brain. Nine separate brain regions (frontal, temporal, occipital, and cerebellar cortices; thalamus; corpus striatum;
자사의 과학자팀은 생명 과학, 재료 과학, 화학 합성, 크로마토그래피, 분석 및 기타 많은 영역을 포함한 모든 과학 분야에 경험이 있습니다..
고객지원팀으로 연락바랍니다.