
추천 제품







생물학적 소스
mouse
Quality Level
항체 형태
purified immunoglobulin
항체 생산 유형
primary antibodies
클론
L12B4, monoclonal
종 반응성
human
반응하면 안 됨
shark, mouse
제조업체/상표
Chemicon®
기술
immunocytochemistry: suitable
immunoprecipitation (IP): suitable
western blot: suitable
동형
IgG2a
특이성
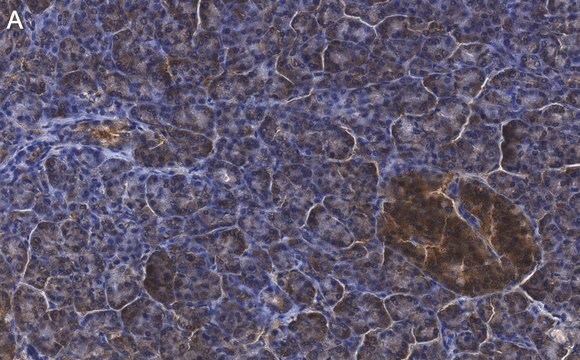
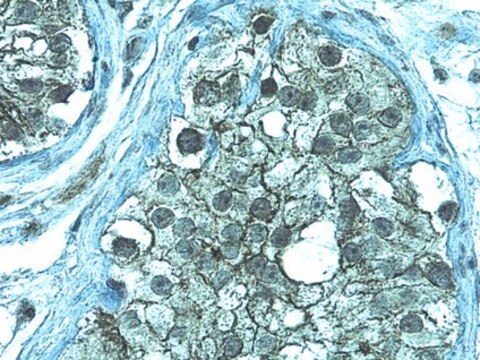
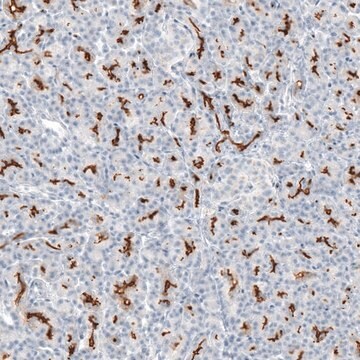
Human cystic fibrosis transmembrane conductance regulator (CFTR). Recognizes an epitope at the cytoplasmic region preceding the first nucleotide binding domain between residues 386 to 412.
Recognizes CFTR, Mr 170kDa and two additional proteins at ~ Mr 100 and 140kDa.
Recognizes CFTR, Mr 170kDa and two additional proteins at ~ Mr 100 and 140kDa.
면역원
Epitope: a.a. 386-412 of human CFTR
애플리케이션
Anti-Cystic Fibrosis Transmembrane Conductance Regulator Antibody, a.a. 386-412, clone L12B4 is an antibody against Cystic Fibrosis Transmembrane Conductance Regulator for use in IC, IP & WB.
Research Category
Neuroscience
Neuroscience
Research Sub Category
Ion Channels & Transporters
Ion Channels & Transporters
Western blot: 1-10μg/mL, note do not boil the lysate, incubate at 80C for 30 minutes prior to running SDS-PAGE. CTFR aggregates upon boiling. Antibody recognizes CFTR at 170kDa and two additional proteins at 100 & 140kDa.
Immunoprecipitation
Immunofluorescence
Note: Does not work on paraffin embedded tissue.
Optimal working dilutions must be determined by the end user.
Immunoprecipitation
Immunofluorescence
Note: Does not work on paraffin embedded tissue.
Optimal working dilutions must be determined by the end user.
물리적 형태
Format: Purified
Purified immunoglobulin. Liquid in 0.02 M Phosphate buffer, 0.25 M NaCl, pH 7.6 with 0.1% sodium azide.
저장 및 안정성
Maintain at 2-8°C in undiluted aliquots up to 6 months.
기타 정보
Concentration: Please refer to the Certificate of Analysis for the lot-specific concentration.
법적 정보
CHEMICON is a registered trademark of Merck KGaA, Darmstadt, Germany
면책조항
Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to humans or animals.
적합한 제품을 찾을 수 없으신가요?
당사의 제품 선택기 도구.을(를) 시도해 보세요.
Storage Class Code
10 - Combustible liquids
WGK
WGK 2
Flash Point (°F)
Not applicable
Flash Point (°C)
Not applicable
시험 성적서(COA)
제품의 로트/배치 번호를 입력하여 시험 성적서(COA)을 검색하십시오. 로트 및 배치 번호는 제품 라벨에 있는 ‘로트’ 또는 ‘배치’라는 용어 뒤에서 찾을 수 있습니다.
Graeme W Carlile et al.
Scientific reports, 12(1), 4595-4595 (2022-03-19)
Most cases of cystic fibrosis (CF) are caused by class 2 mutations in the cystic fibrosis transmembrane regulator (CFTR). These proteins preserve some channel function but are retained in the endoplasmic reticulum (ER). Partial rescue of the most common CFTR
Celeste Riepe et al.
Molecular biology of the cell, 35(2), ar15-ar15 (2023-11-29)
Over 80% of people with cystic fibrosis (CF) carry the F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR), a chloride ion channel at the apical plasma membrane (PM) of epithelial cells. F508del impairs CFTR folding causing it to
Mauricio Di Fulvio et al.
PloS one, 15(12), e0242749-e0242749 (2020-12-03)
Cystic fibrosis (CF) is due to mutations in the CF-transmembrane conductance regulator (CFTR) and CF-related diabetes (CFRD) is its most common co-morbidity, affecting ~50% of all CF patients, significantly influencing pulmonary function and longevity. Yet, the complex pathogenesis of CFRD
Chiara Brandas et al.
Biomolecules, 11(10) (2021-10-24)
Cystic fibrosis (CF) is caused by loss-of-function mutations in the CF transmembrane conductance regulator (CFTR) protein, an anion channel that regulates epithelial surface fluid secretion. The deletion of phenylalanine at position 508 (F508del) is the most common CFTR mutation. F508del
Javier Sanz et al.
European journal of human genetics : EJHG, 18(2), 212-217 (2009-09-03)
Cystic fibrosis (CF) is one of the most common genetic diseases in the Caucasian population and is characterized by chronic obstructive pulmonary disease, exocrine pancreatic insufficiency, and elevation of sodium and chloride concentrations in the sweat and infertility in men.
문서
16HBE14o- human bronchial epithelial cells used to model respiratory epithelium for the research of cystic fibrosis, viral pulmonary pathology (SARS-CoV), asthma, COPD, effects of smoking and air pollution. See over 5k publications.
자사의 과학자팀은 생명 과학, 재료 과학, 화학 합성, 크로마토그래피, 분석 및 기타 많은 영역을 포함한 모든 과학 분야에 경험이 있습니다..
고객지원팀으로 연락바랍니다.