
추천 제품
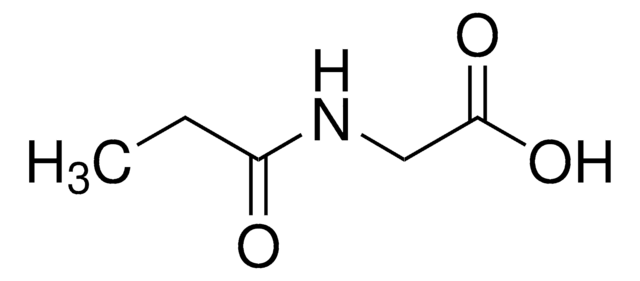





Grade
analytical standard
Quality Level
분석
≥98.0% (HPLC)
유통기한
limited shelf life, expiry date on the label
응용 분야
clinical testing
형식
neat
저장 온도
2-8°C
InChI
1S/C7H13NO3/c1-3-5(2)7(11)8-4-6(9)10/h5H,3-4H2,1-2H3,(H,8,11)(H,9,10)
InChI key
HOACIBQKYRHBOW-UHFFFAOYSA-N
생화학적/생리학적 작용
2-Methylbutyrylglycine is an acyl glycine. Acyl glycines are normally minor metabolites of fatty acids. However, the excretion of certain acyl glycines is increased in several inborn errors of metabolism. In certain cases the measurement of these metabolites in body fluids can be used to diagnose disorders associated with mitochondrial fatty acid beta-oxidation. Acyl glycines are produced through the action of glycine N-acyltransferase (EC 2.3.1.13) which is an enzyme that catalyzes the chemical reaction:. acyl-CoA + glycine ↔ CoA + N-acylglycine. The isolated excretion of high levels of 2-methylbutyrylglycine (2-MBG) is the hallmark of short/branched-chain acyl-CoA dehydrogenase deficiency or SBCADD. The disorder is also called 2-methylbutyryl-CoA dehydrogenase deficiency and has been associated with autism and mental retardation. SBCADD is a recently described autosomal recessive disorder caused by a defect in the degradation pathway of L- isoleucine leading to increased urinary excretion of 2-methylbutyryl glycine. The enzymatic defect results from disruption of the SBCAD gene. Deficiency of SBCAD leads to accumulation of its substrate, 2-methylbutyryl-CoA within the mitochondrion. This substance is transesterified with glycine by the mitochondrial enzyme acyl-CoA glycine-N-acyltransferase (glycine-N-acylase) to form 2-methylbutyryl glycine. Affected patients can be divided into two categories. The first category consists of infants detected by newborn screening programs. These infants are treated with diet and remain without clinical symptoms. In the second category affected patients are diagnosed because they presented clinically with seizures and psychomotor delay and have increased urinary excretion of 2-methylbutyryl glycine. 2-Methylbutyrylglycine has also been found in the urine of patients with propionyl-CoA carboxylase deficiency after consuming isoleucine. 2-methylbutyrylglycine is also elevated in the urine of patients with glutaric aciduria II and ethylmalonic encephalopathy.

신호어
Warning
유해 및 위험 성명서
Hazard Classifications
Eye Irrit. 2 - Skin Irrit. 2
Storage Class Code
11 - Combustible Solids
WGK
WGK 3
Flash Point (°F)
Not applicable
Flash Point (°C)
Not applicable
Oivind J Kanavin et al.
Journal of medical case reports, 1, 98-98 (2007-09-22)
2-methylbutyryl-CoA dehydrogenase deficiency or short/branched chain acyl-CoA dehydrogenase deficiency (SBCADD) is caused by a defect in the degradation pathway of the amino acid L-isoleucine. We report a four-year-old mentally retarded Somali boy with autism and a history of seizures, who
Stanley H Korman et al.
Clinical chemistry, 51(3), 610-617 (2004-12-24)
Isolated excretion of 2-methylbutyrylglycine (2-MBG) is the hallmark of short/branched-chain acyl-CoA dehydrogenase deficiency (SBCADD), a recently identified defect in the proximal pathway of L-isoleucine oxidation. SBCADD might be underdiagnosed because detection and recognition of urine acylglycines is problematic. Excretion of
L Sweetman et al.
Biomedical mass spectrometry, 5(3), 198-207 (1978-03-01)
A number of previously unrecognized abnormal metabolites have been identified and quantitated in the urine of a patient with an inherited deficiency of propionyl-CoA carboxylase. These included the isoleucine metabolites 2-methyl-3-hydroxybutyric acid and 2-methylacetoacetic acid. These isomers 3-hydroxyvaleric acid and
자사의 과학자팀은 생명 과학, 재료 과학, 화학 합성, 크로마토그래피, 분석 및 기타 많은 영역을 포함한 모든 과학 분야에 경험이 있습니다..
고객지원팀으로 연락바랍니다.